
- Summary
- Introduction
- What is a Clinical Trial Protocol?
- Foundations of a Clinical Trial Protocol
- Steps in Clinical Trial Protocol Development
- Roles and Responsibilities in Protocol Development
- Ethical and Regulatory Considerations
- Challenges and Pitfalls in Protocol Design
- The Rise of eProtocols
- How AI is Advancing eProtocol Design
- Best Practices and Checklist for Strong Protocol Design
- Conclusion
Summary
A clinical trial protocol is the foundation of clinical research, detailing objectives, study design, eligibility, safety oversight, and data analysis. Its structure ensures consistency and credibility, guiding investigators while protecting participants and maintaining scientific rigor. With digital solutions, eProtocols and AI are modernizing protocols in clinical trials, making processes more adaptive and reducing inefficiencies.
Introduction
Every clinical trial depends on a detailed plan that outlines how the study will be run, how participants will be safeguarded, and how results will be measured. This plan, called a clinical trial protocol, is the foundation of research integrity.
Industry research shows that nearly three out of four clinical trial protocols require at least one amendment after initiation, often adding months of delays and significant costs. A well-designed protocol minimizes these risks by aligning scientific goals, ethical standards, and operational realities.
What is a Clinical Trial Protocol?
A clinical trial protocol is a comprehensive document that outlines how a clinical research study will be conducted. It details the study objectives, trial design, participant eligibility criteria, methods for data collection, safety monitoring procedures, and the statistical plan for analysis. In essence, it is the rulebook that guides investigators, protects participants, and ensures the data collected is scientifically valid.
The protocol also serves multiple functions:
- Operational Guide: Directs investigators and site staff to run the trial consistently across all sites.
- Ethical Safeguard: Explains how participant rights, safety, and confidentiality will be protected.
- Scientific Framework: Provides the rationale, objectives, and methodology that give the study credibility.
- Regulatory Document: Demonstrates compliance with ethics committees and regulatory authorities.
Without a protocol, a trial risks becoming disorganized and losing the consistency needed for valid results. With one, every stakeholder has a common reference point that defines expectations and responsibilities.
Foundations of a Clinical Trial Protocol
Protocols may differ depending on the type of study, but certain core elements are always included to ensure safety and scientific integrity. Below are the core building blocks.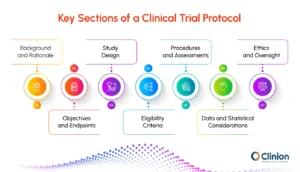
1. Background and Rationale
Every protocol begins with context: what is known about the study focus, what data exists for the investigational product, and why this particular study is necessary. This section also describes the expected benefits and potential risks, ensuring the rationale for moving forward is scientifically and ethically sound.
2. Objectives and Endpoints
Objectives define what the study aims to achieve, while endpoints explain how success will be measured.
- Primary objectives are the main goals, such as proving efficacy or establishing safety.
- Secondary and exploratory objectives provide supporting evidence, often focusing on additional outcomes such as quality of life or patient-reported measures.
Clear, measurable endpoints prevent ambiguity later when regulators and stakeholders evaluate results.
3. Study Design
This is the blueprint of the trial. It specifies the type of study (randomized controlled, crossover, adaptive), how participants are assigned to groups, whether blinding is used, and the flow of activities across visits. Well-designed studies anticipate bias and put safeguards in place to minimize it.
4. Eligibility Criteria
Inclusion and exclusion criteria define who can take part. These rules strike a balance between protecting participants, ensuring the population is appropriate, and allowing for feasible recruitment. For example, overly narrow criteria may improve internal validity but can make enrollment unreasonably slow.
5. Procedures and Assessments
This section details exactly what will happen at each stage: interventions, diagnostic tests, imaging, questionnaires, and follow-up assessments. Many protocols include a schedule of events table that maps assessments against timepoints for clarity.
6. Data and Statistical Considerations
A protocol must explain how data will be collected, managed, and analyzed. Statistical methods, handling of missing data, and plans for interim analyses are all included here. This ensures transparency and reduces disputes about interpretation later.
7. Ethics and Oversight
Protocols explain how informed consent will be obtained, how participant privacy will be safeguarded, and how monitoring boards or ethics committees will oversee the trial. Independent oversight provides credibility and reinforces participant trust.
Steps in Clinical Trial Protocol Development
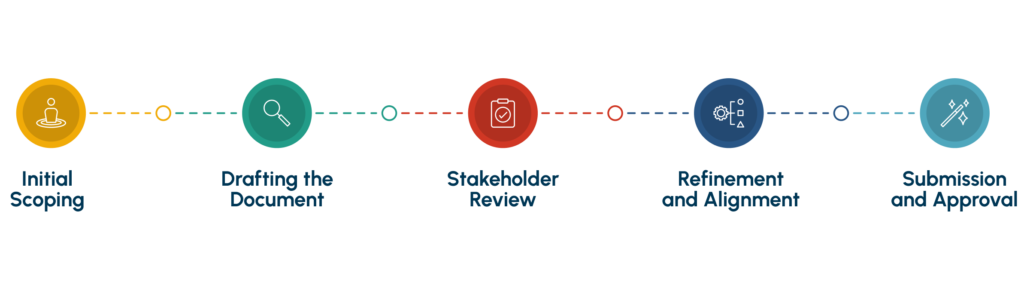
Initial Scoping
- Define the primary research question the study is designed to answer.
- Assess feasibility based on resources, timelines, target population, and site capabilities.
- Identify potential challenges early to reduce the likelihood of amendments later.
Drafting the Document
- Sections are authored collaboratively by sponsors, clinical experts, statisticians, and medical writers.
- Patient advocates may also be consulted to highlight practical concerns that affect participation.
- Standardized templates are often used to ensure consistency and that no critical content is overlooked.
Stakeholder Review
- Drafts are circulated among investigators, site staff, data managers, and regulatory specialists.
- Their input helps identify operational blind spots such as scheduling conflicts or unclear safety procedures.
- This collaborative review stage ensures the protocol is both scientifically valid and logistically feasible
Refinement and Alignment
- Revisions are made to address feedback and align scientific goals with operational realities.
- This stage helps prevent protocol amendments during the trial, which can otherwise delay timelines and increase costs.
Submission and Approval
- The final protocol is submitted to ethics committees, institutional review boards (IRBs), and regulatory authorities.
- Approvals are mandatory before participant recruitment can begin, making this the final checkpoint before trial launch.
Roles and Responsibilities in Protocol Development
Developing a clinical trial protocol is a collaborative effort that brings together experts from different disciplines. Each stakeholder contributes specific knowledge that shapes the quality and credibility of the final document.
Sponsors:
Provide funding and overall oversight, ensuring the protocol aligns with strategic goals and regulatory requirements.
Principal Investigators (PIs):
Responsible for implementing the protocol at study sites and ensuring participant safety throughout the trial.
Clinical Research Coordinators (CRCs):
Manage day-to-day trial operations, including scheduling, participant communication, and data entry and documentation.
Statisticians and Data Managers:
Design and validate the analysis plan so that it is rigorous and data handling methods are robust.
Medical Writers:
Translate complex scientific input into a clear, consistent document.
IRBs/Ethics Committees:
Independently evaluate the protocol to safeguard participant rights and monitor compliance with Good Clinical Practice (GCP).
This multidisciplinary approach reduces blind spots and ensures protocols are both rigorous and feasible.
Ethical and Regulatory Considerations
Ethics and regulation form the foundation of every clinical trial. A protocol must demonstrate how the study will safeguard participants and meet established standards for clinical research.
Informed Consent:
Participants must be given clear, accessible information about the trial, including potential risks and benefits, so they can make voluntary decisions about participation.
Participant Privacy:
Protocols should outline how personal and medical data will be protected, with compliance to standards such as HIPAA and GDPR, where applicable.
Independent Oversight:
IRBs or ethics committees review the protocol to ensure that studies are justified, ethically acceptable, and monitored throughout.
Regulatory Compliance:
Protocols must follow GCP guidelines as well as local and international regulations.
By addressing these areas, ethical oversight helps protect individuals taking part in the trial and reinforces the credibility of the study.
Challenges and Pitfalls in Protocol Design
Even with thorough planning, clinical trial protocols often face practical challenges that can disrupt study timelines and data quality. Some of the most common issues include:
Frequent Amendments:
Often the result of unclear objectives or gaps in protocol detail, leading to delays and additional review cycles.
Overly Complex Designs:
Complicated visit schedules and testing requirements can make trials harder to manage and discourage participation.
Protocol Deviations:
Occur when procedures are not followed exactly, either due to oversight or participant non-compliance.
Recruitment Barriers:
Narrow eligibility criteria can limit the number of potential participants.
Example: How Pitfalls Affect Trials
Challenge | Consequence | Solution |
Unclear endpoints | Disputes in interpretation | Define endpoints using validated and measurable tools |
Excessive visit schedules | Participant fatigue, dropouts | Streamline procedures where feasible |
Inflexible wording | High risk of deviations | Allow limited flexibility in non-critical areas |
Narrow eligibility | Delayed recruitment | Broaden inclusion criteria while ensuring safety |
Participant Perspective
While protocols are technical documents, they directly impact participants’ experiences. The number of visits, frequency of tests, and reporting of side effects are all governed by the protocol. If not designed with participants in mind, even the most scientifically rigorous trial may struggle with recruitment and retention.
Increasingly, patient advocates are involved in protocol development to provide input on feasibility and acceptability. This shift helps ensure that protocols balance scientific needs with participant well-being.
The Rise of eProtocols
Traditional clinical trial protocols are static Word or PDF documents. While they serve as the reference point for trial conduct, they are difficult to update, prone to version control issues, and often require manual re-entry of information into multiple systems.
An eProtocol is an electronically generated version of the clinical trial protocol. By digitizing protocol creation, research teams can collaborate more effectively, maintain stronger version control, and work with greater operational efficiency.
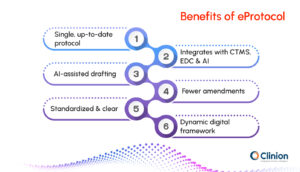
Key advantages of eProtocols include:
- Centralized access to a single, up-to-date version of the protocol.
- Interoperability with systems like CTMS, EDC, and regulatory submission tools.
- The ability to reduce protocol amendments through real-time collaboration and standardized formats.
Digitization moves protocols from being a document that must be interpreted to a dynamic framework that integrates with modern trial technology.
How AI is Advancing eProtocol Design
Artificial intelligence has already reshaped how protocols are created and managed. Tasks that once required weeks of drafting, review, and alignment can now be completed far more quickly with AI-driven tools.
AI applications include:
- Using validated templates that meet regulatory requirements to generate compliant protocol drafts.
- Producing structured drafts from a short synopsis in a fraction of the time.
- Drawing on historical trial data to refine eligibility criteria, endpoints, and operational feasibility.
- Enabling real-time collaboration and version control so teams remain aligned across global sites.
Looking ahead, agentic AI represents the next wave. These systems are designed not only to generate content but also to take initiative, simulate trial outcomes, and refine protocols dynamically as new information becomes available. AI is already established in automating protocol development, but agentic AI is expected to extend this further by supporting proactive decision-making throughout the trial lifecycle.
Best Practices and Checklist for Strong Protocol Design
Developing a protocol that works in practice requires careful planning and attention to both scientific and operational detail. Researchers can use the following checklist as a guide:
- Define primary and secondary objectives clearly.
- Align endpoints with outcomes that can be measured and validated.
- Keep eligibility criteria realistic to support participant recruitment.
- Provide procedures in sufficient detail for consistent application across sites.
- Anticipate operational challenges and address them early.
- Establish processes for data integrity and quality control.
- Ensure compliance with ethical and regulatory standards.
- Seek input from stakeholders during the design phase rather than after.
Conclusion
The clinical trial protocol remains the foundation of research, shaping how studies are conducted and how participants are protected. Poorly designed protocols can cause delays and compromise data quality, whereas strong ones provide the clarity and reliability needed by regulators, investigators, and sponsors.
As trials become more complex, digital innovation is reshaping how protocols are created and managed. eProtocols and AI are already improving speed and consistency, while the coming wave of agentic AI is expected to make protocol development more adaptive and intelligent than ever before.
By following established best practices and embracing innovation thoughtfully, research teams can ensure their protocols continue to serve as effective frameworks for safe and credible clinical trials.


